YOU ARE HERE: Research Design & Methods
Research Design & Methods
- Design Criteria for Libraries
- Summary of Library Members
- Yields and Delivered Sample Sizes
- Second Generation Libraries
- Sample Handling During Synthesis
- Compound Purification and Characterization of Library Members
- Sample Storage and Archival of Analytical Data
- Public Dissemination
- Timeline of the Proposed Work
- Project Management
General Features
A compound library for lead discovery should optimally sample a range of topographical space with a variety of pharmacophores, since this provides the greatest opportunity for identifying a complementary interaction in the biomolecular recognition event (Gorse, 2000)*.
Generally the size and shape of a compound library will dominate the relative activity of the compound class against a single target, with outliers being those compounds that form additional electrostatic or hydrophobic interactions or both, as illustrated in Figure 11.
If the compounds form a common binding interaction or have a common scaffold, it is sometimes possible to discern the important and favorable contacts by a 3D-QSAR comparison of the structures.
Indeed, the shape-property analysis of compounds has been given renewed attention recently (Carpy, 2003; Hofbauer, 2004; Klein, 2002; Mason, 1999b; Polanski, 2000; Polanski, 2003)*.
The selection criterion for the compounds in each class of this proposal is discussed in the appropriate section below.
Back to Design of Library Navigation
Back to the Top
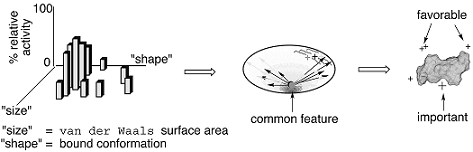
Figure 11. Illustration describing how size and shape can predict the relative bioactivity of many compounds within a given structural class and how, if there is a common binding element or mode, the outliers can be expected to form favorable hydrophobic or electrostatic interactions or both when bound (Gorse, 2000)*.
Building Blocks for I45DC, I45EA, and I45DE
The compounds to be synthesized in this aim are expected to be the most useful for targeting kinases, G-protein coupled receptors, ion channels, cytoskeletal proteins, and enzymes. These library members have a comparably narrow size range. On the other hand, the substituents in these classes can dramatically influence the preferred conformation of the scaffold, and thereby influence the relative orientation of the pharmacophores. The value of these libraries will be realized in the relative activity of the series, as this will provide detailed SAR for a given target, as illustrated with the antiproliferative compounds against HL-60 cells (Perchellet, 2005)*. This information will then be used to design more potent and selective analogs, either one at a time or in small parallel libraries. Our chosen substituents for these series are
different enough from one another to not duplicate efforts in discovery, but to rather focus on identifying a lead structure. The selected alkanamines, heterocyclic amines, anilines, amino acid esters, and alcohols for use in this specific aim can be viewed here.
Back to Specific Aim 1 Navigation
Back to the Top
Synthetic Methods for I45DC, I45EA, and I45DE
The methodology to synthesize the vast majority of these library members is detailed in the preliminary results (see Synthesis of I45DA Derivatives). It is easy to isolate the intermediate pyrazines from the reaction of 1.2 with secondary alkanamines, anilines, amino acid esters, and alcohols. Thus, I45DA derivatives bearing these substituents become straightforward to prepare and the purification protocol for the product from a subsequent reaction will yield analytically clean product (> 95% purity) with significant reliability.
Symmetrically N,N'-disubstituted I45DC
A total of 22 compounds will be delivered in 50-100 mg quantities. The primary alkanamines, secondary alkanamines, and amino acids esters will be used for the preparation of these compounds.
Back to Specific Aim 1 Navigation
Back to the Top
Dissymmetrically N,N'-disubstituted I45DC
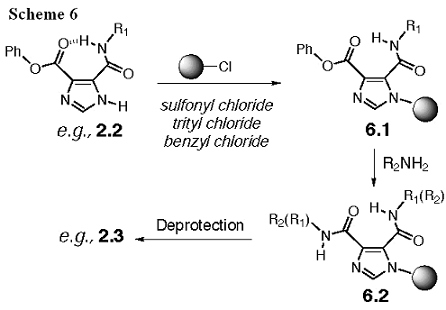
The delivered amounts for the 544 compounds in this class will be 20-50 mg. The primary alkanamines are too reactive at the acyl imidazole bond to be selective. Thus, the dI45DCs with alkanamine substituents represent the greatest synthetic challenge in this aim. On the other hand, we have improved the synthetic methods to these derivatives, as described in I45DCs Substituted with Alkanamines (Wiznycia, 2002)*. These reactions can be done in parallel, though the reactions may require more attention during the purification phase. This is not an extreme burden to the project, since a total of only 36 unique combinations exist, and we could use preparative HPLC in order to purify each member. An alternative synthetic strategy, if necessary, is to secure intermediates 2.2 to solid phase resins through imidazole ring alkylation to give 6.1 (Scheme 6). The phenyl ester can then be substituted with a primary alkanamine to yield 6.2, before deprotection to give 2.3.
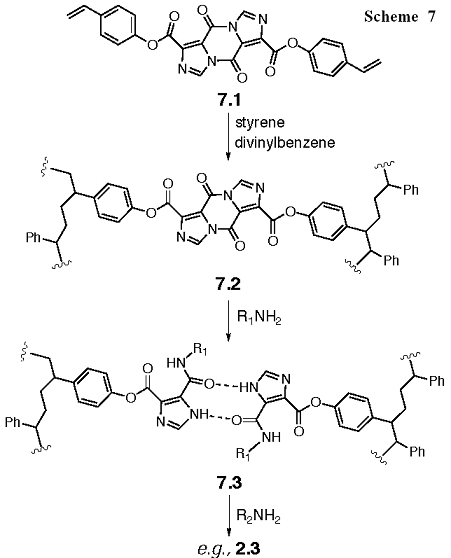
A yet untested but exciting way to perform the solid-phase synthesis would be to prepare our own resin, and this will be pursued as an option to the above synthesis of dI45DCs as necessary. A pyrazine containing a vinyl phenol, 7.1, would be included with styrene and divinylbenzene in a polymerization reaction to yield 7.2 (Scheme 7). This resin can be reacted with an alkanamine to give 7.3 followed by a second alkanamine to yield products such as 2.3 that will simply wash from the resin for easy recovery. The level of cross-linking and other important parameters would be explored (Vaino, 2000)* in order to optimize the reaction. The other variations of these I45DCs that we will provide include the following: secondary alkanamines-primary alkanamines (27 cmpds), heterocyclic amines-primary alkanamines (27 cmpds), heterocyclic amines-secondary alkanamine (9 cmpds), heterocyclic amines-amino acid esters (63 cmpds), anilines-primary alkanamines (81 cmpds), anilines-secondary alkanamines (27 cmpds), anilines-amino acid esters (90 cmpds), amino acid esters-primary alkanamines (90 cmpds), amino acid esters-secondary alkanamines (30 cmpds), and amino acid esters-amino acid esters (100 cmpds).
The order of the substituents in the dI45DC libraries mentioned above is an indication of which nucleophile will be added to 1.2 in order to yield the corresponding pyrazine intermediate. In general,
anilines and heterocyclic amines are the easiest to prepare and purify for the next step (simple vacuum filtration followed by washing of the solid with solvent), so this is the preferred option whenever possible. An example of one such synthesis is shown in Scheme 4. It is possible to switch the reaction order of substituents in some cases, and we would do so if it is necessary to either improve yields or simplify purification. We note that some of the compounds will serve as intermediates for libraries in specific aim 2. These compounds will be prepared in greater amounts on an individual or parallel basis for those purposes.
Back to Specific Aim 1 Navigation
Back to the Top
I45EA
A total of 132 compounds will be delivered in 20-50 mg quantities. The compounds in this class are described as follows: alcohols-primary alkanamines (54 cmpds), alcohols-secondary alkanamines (18 cmpds), alcohols-amino acid esters (60 cmpds).
Back to Specific Aim 1 Navigation
Back to the Top
I45DE
A total of 21 compounds will be delivered in 20-50 mg quantities, which includes the symmetrically disubstituted and unique dissymmetrically disubstituted compounds. We have not prepared dissymmetric I45DEs to date, but expect their synthesis to be straightforward since we can prepared pyrazine esters (Baures, 2002; Wiznycia, 2002)*. The acyl imidazole bond in pyrazine esters will be sufficiently reactive to form an ester with the primary and secondary alcohols in our list of building blocks, though catalytic additives to promote product formation, such as dimethylaminopyridine (DMAP), can be used as necessary. Whenever possible, the most hindered alcohol will be used in the synthesis first. In this way the potential for ester exchange leading to symmetric I45DEs as an impurity in the reaction can be minimized.
Back to Specific Aim 1 Navigation
Back to the Top
Ring-alkylated I45DC
Select products from specific aim 1 will be derivatized on the imidazole ring with methyl iodide and benzyl chloride by the method shown previously in Ring-Alkylated I45DCs. The compounds selected for this purpose will be the I45DCs symmetrically substituted with primary alkanamines, secondary alkanamines, and amino acid esters (22 compounds yielding 44 derivatives in this aim). These derivatives will be delivered in 20-50 mg quantities.
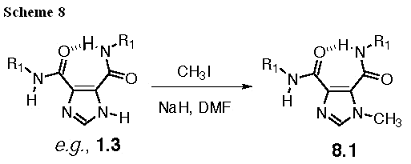
The synthesis of a benzyl derivative is shown in Scheme 5 (Ring-Alkylated I45DCs). We have not yet attempted the methyl substitution of the ring. However, there is a reported method for ring methylation of I45EAs that uses methyl iodide with sodium ethoxide in ethanol at 50°C (Yasuda, 1987)*. This reported method will be used if necessary, although this would not be ideal for the amino acid esters, since ester exchange can occur for the benzyl esters. A variation on this method will be to use sodium hydride in dimethylformamide in order to create the imidazole anion (the most acidic hydrogen) in the presence of methyl iodide, thus excluding the alcohol that could exchange with the esters (Scheme 8). In addition, we note that I45DEs have been ring alkylated with sulfonates (Chen, 2001)*, orthoformates (Collman, 2000)*, bromoalkanes (Chen, 2000)*, and benzylic halides (Anderson, 1989)*. All of these methods are examples of the synthetic chemistry that can yield these derivatives.
Alkylation of the imidazole ring in dI45DCs can lead to mixtures of the two possible products, and this is anticipated if the ring is derivatized with a probe for a bioassay. The biologically active compounds identified in this manner will be valuable tools, although separation of the two similar products may be difficult in some cases. We will select 20 dI45DCs from the following classes: aniline-secondary alkanamine, aniline-amino acid ester, and I45EAs substituted with amino acids. The reason for choosing these compounds is because they give disparate conformations depending upon the product of the alkylation. Thus, alkylation of the imidazole on the side of the intramolecular hydrogen bond donor yields a product that still forms this hydrogen bond, whereas alkylation on the side of the intramolecular hydrogen bond acceptor precludes intramolecular hydrogen bond formation (Rush, 2005; Yasuda, 1987)*.
If both products are formed in the reaction, the different properties for the isomers will allow us to separate them. This was the case in a previous example (Yasuda, 1987)*. In such cases, both products will be delivered if their amounts are sufficient. We will also determine and report the product distribution for each of the reactions, as this will be valuable for others planning ring alkylations with a probe, as well as in interpreting the results if the products are used as mixtures.
Back to Specific Aim 2 Navigation
Back to the Top
Deprotected Monomeric I45DC
A total of 214 I45DA derivatives described in specific aim 1 contain a single protected amine and are quite distinct from one another (only lysine benzyl ester and not lysine tert-butyl ester derivatives were considered). For example, there are 4 primary alkanamines with a protected amine, and in combination gives 8 unique products with secondary alkanamine building blocks not bearing a protected nitrogen. Amines were chosen for the free functional groups because so many probes are already commercially available for reacting with amines.
We will deprotect these 214 combinations with 4 N HCl in dioxane in order to give the HCl salts following lyophilization. Analysis by LC-MS will be done to ensure the lack of side products. Scavengers will be used as necessary or when sensitive functionalities are present. Side products like trityl chloride can be removed from the product salt by selective solubilization (washing).
Likewise, there are 108 unique amino acid tert-butyl ester I45DA derivatives where no other functionalities sensitive to deprotection conditions are present. These will be deprotected to yield the free carboxylic acids following lyophilization. This is an alternate set of compounds for derivatization through a carboxylic acid.
The 322 total deprotected I45DA derivatives will be useful for researchers needing to use compounds with probes in their bioassay.
Back to Specific Aim 2 Navigation
Back to the Top
Substituent Modified I45DC
This specific aim also describes substituent modified analogs. Many of the building blocks selected for this project can be appended with reactive functional groups such as alcohols, amines, and carboxylic acids.
These variations would be used for preparing tagged versions of biologically active compounds identified through the Molecular Libraries Screening Center Network (MLSCN). While we hope new I45DA derivatives are identified for such development sometime during the second year of the grant, we would begin tagging the I45DCs shown to be antiproliferative to HL-60 cells (see Antiproliferative I45DCs: Potential Kinase Inhibitors) if this is not the case. For the dI45DCs in this work, the tags would be appended to the aniline positions (para to the amide), which is a region that showed little bioactivity change even when rather large substituents were incorporated here. We reason that these sites are either in solvent accessible regions or that a large pocket exists at this location when the dI45DC is bound. Thus, we anticipate covalently
bound probes at this location will not adversely affect the binding of the compounds to their target(s).
Back to Specific Aim 2 Navigation
Back to the Top
Known Bis-I45DC
The bis-I45DCs described in the preliminary results (see Protein-Protein Interaction Inhibitors) were designed to mimic an α-helix of the large extracellular loop of tetraspanin CD81 (VanCompernolle, 2003)*. These specific compounds and their application to treating Hepatitis C infections is the property of Kansas State University Research Foundation. For this reason and per the RFA instructions that prefers no patents either exist or are pending for the submitted libraries (or will be pursued in the future), we will not pursue this specific class in this proposal.
Back to Specific Aim 3 Navigation
Back to the Top
General Design Considerations of Oligomeric I45DC
There is reason to think that other designs of bis-I45DCs could yield bioactive compounds for studying the cell. The unique designs proposed herein use substituents other than amino acids to decorate the bis-I45DCs, expand the scaffold to include tris-I45DCs, and include a series of unique oligomeric I45DCs with amino acid substituents. We think that compounds in this class of derivatives could bind to kinases, receptors, channels, or cytoskeletal proteins-like earlier I45DA derivatives in this proposal-or they might inhibit a variety of protein-protein interactions in the cell.
To consider the potential for bioactivity with these compounds, one needs to only examine the recent literature, as there are known symmetrical small molecules (bis structures) that have interesting biological activity. For example, suramin has a diaryl urea core and inhibits several important protein-tyrosine phosphatases (PTPs) in the cell (McCain, 2004)*. Suramin analogs are also potent antagonists at purine receptors P2X (Hulsmann, 2003)*, and were recently used to gather evidence for P2X receptor dimers (Nicke, 2005)*. This later bioactivity is particularly interesting, as G-protein coupled receptor dimers, as well as larger aggregates, may represent the dominant form of these receptors in the cell (Javitch, 2003; Milligan, 2004)*. Thus, small molecule tools with selectivity toward these targets would be helpful in characterizing these assemblies.
We also think the oligomeric I45DCs compounds can target protein-protein interactions. These are traditionally difficult targets, yet the interactions are subject to the same factors governing any chemical process, such as kinetics, thermodynamics, concentration of the proteins, the context of the interaction, and even the time course of the overall binding event (Janin, 2000; Ma, 2001). It is therefore important to consider the features of known interfaces between proteins for guidance in designing potential inhibitors.
Perhaps a surprise to some, protein-protein interfaces do not solely bury hydrophobic surfaces, as polar residues are also found in interfacial "hot spots" (Ma, 2001)*. A comparison of globular proteins with protein-protein interfaces show that the two have similar amino acid compositions (Lo Conte, 1999)*. The amino acid residues favored in antibody-antigen interactions, which are dominated by side chain-side chain or side chain-main chain interactions, are Tyr > Asp > Asn > Ser > Glu > Trp, although Arg and Lys are also amino acids that can significantly stabilize the interaction (Jackson, 1999)*.
Proteins involved in signal transduction generally bury a large amount (= 550 Å2 per protein) of surface area (Janin, 2000)*. Some proteins are quite plastic and capable of varying their interface conformation to coincide with the interaction (Ma, 2001)*. The necessary features for inhibiting protein-protein interactions by small molecules-particularly pharmacophoric spacial matching-have been addressed by several groups (Burgess, 2001; Cochran, 2000; McCormick, 2000; Toogood, 2002; Zutshi, 1998)*, and the need for conformational flexibility in drug design has also been highlighted recently (Bursavich, 2002)*. These factors are all relevant to addressing the complementarity of interfaces between proteins and non-natural partners (Siemion, 2004)*.
We think the ability to easily functionalize I45DA with amino acids and have separation distances comparable to known protein secondary structure is but one advantage of this scaffold. An added feature is the conformational plasticity of the intramolecular
hydrogen bond, making this scaffold valuable in the search for inhibitors of protein-protein interactions.
Back to Specific Aim 3 Navigation
Back to the Top
Structures and Synthesis of Bis- and Tris-I45DC
The preparation of bis- and tris-I45DCs will be straightforward based on our preliminary results (Bis-I45DCs). Thus, aniline pyrazines 4.1 will be opened with a series of diamines and one triamine to give the products represented by 9.1 in Scheme 9.
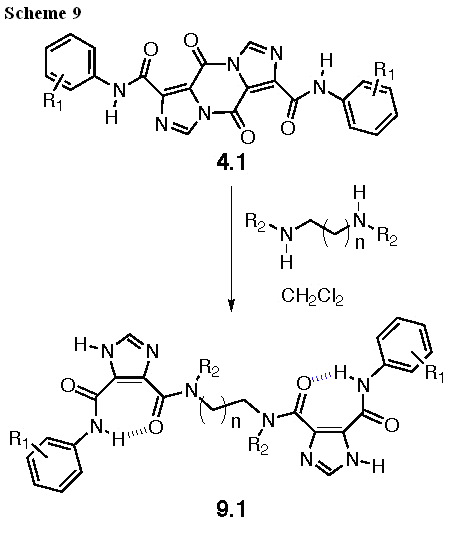
The diamines and triamine to be used for these libraries are shown in Figure 13. These linkers were selected to vary length, general overall conformation, partial side chain mimicry by N-ethyl and N-benzyl substituents, and the number of appended I45DCs. In all, a total of 63 compounds in 20-50 mg quantities will be provided with greater than 95% purity as determined by LC-MS with UV detection at 214 nm. To accomplish this, we will use commercial solid-phase extraction columns for the purification of all library members, rather than prepare them ourselves as described in the preliminary results (see Oligomer Library Synthesis and Purification: A Model Scale-Up). In the unlikely event that this chromatography procedure yields any of the final compounds with less than 95% purity, semi-preparative HPLC will be used to purify the compound(s).
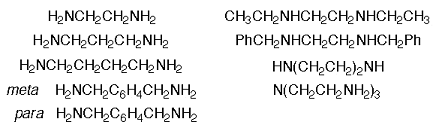
Figure 13. Structures of the diamines and triamine for use in specific aim 3.
Back to Specific Aim 3 Navigation
Back to the Top
Oligomeric I45DC Design
We have also targeted a new class of linear oligomers, based on amino acid substituted I45DCs, that will be useful in identifying inhibitors of protein-protein interactions. In addition to side chain presentation, the compounds have conformational flexibility retained in the amino acid bonds, as well as in the shallow potential energy well resulting from the intramolecular hydrogen bond of the scaffold. Moreover, the compounds have a size sufficient for interfering with protein-protein interactions. The amino acids selected for the oligomer syntheses are based on the residues found in antibody-antigen interactions. While these interfaces are not necessarily the targets of this class of structures, we think these side chains are also likely to contribute significantly at other protein-protein interfaces, and our choices therefore represent a reasonable, albeit minimal, set of residues for this application.
A generic oligomer structure and an indication of the appended amino acids are both given in Figure 14. These amino acids also achieve both a balance of hydrophobic-hydrophilic character and allow for charge-assisted interactions to be recognized at the interface. All of the amino acids with stereogenic centers will be of natural L-configuration.
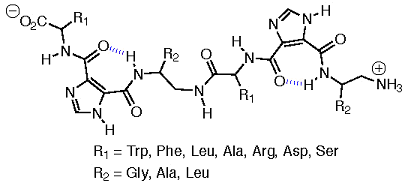
Figure 14. Generic oligomeric structure and targeted amino acids.
Overall Synthesis of Oligomeric I45DCs
The generic synthesis of these oligomers is shown in Scheme 10. One monomer, 10.1, will be a free amine with a tert-butyl ester protected carboxylic acid, whereas the other monomer, 10.2, will be a free acid and tert-butoxycarbonyl (Boc) protected amine. In the case of monomers with Trp, Arg, Asp, and Ser side chains, these will also have Boc, tert-butyl ester, or tert-butyl ether protection of the side chain functionalities. The Trp, Arg, and Ser derivatives are all available commercially. The Asp(OtBu)OBzl will be synthesized from FmocAsp(OtBu)OPfp (Pfp = pentafluorophenol) by addition of benzyl alcohol and subsequent deprotection of the Fmoc protecting group with piperidine in dimethylformamide (DMF).
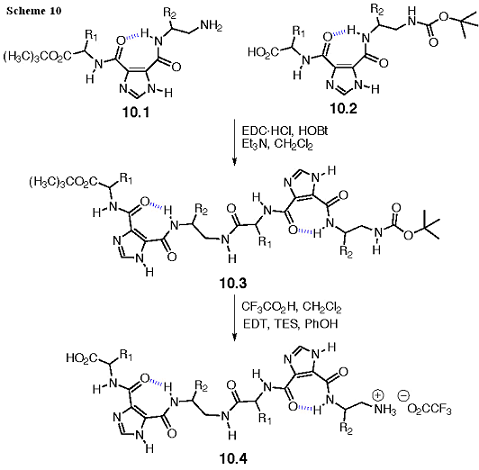
The I45DC monomers 10.1 and 10.2 can be coupled under standard conditions with a water-soluble carbodiimide to provide 10.3. These conditions give good yields and, equally important, the coupled products can be obtained in high yield and purity (> 95%) by extraction. The Tecan Genesis RSP 100 Combinatorial Synthesizer is capable of both the synthesis and extraction. This instrument has four robotic probes with interchangeable septa piercing co-axial (for inert gases) needles and/or disposable tips. Both tip types are conducting and therefore have the capability to do on deck extractions of aqueous/organic phases. We would be prepared to use solid-phase extraction on silica gel in order to achieve this level of purity in the event of a difficult case. In this unlikely scenario, vacuum manifolds will assist in the rapid elution of library members through the solid-phase extraction columns.
Deprotection with trifluoroacetic acid, in the presence of scavengers to include ethanedithiol, triethylsilane, and phenol, will provide the desired oligomers 10.4. Alternatively, 4 N HCl in dioxane can be used for deprotection. Products 10.4 would be more challenging to purify than the protected intermediates, so our focus will be to start with intermediates 10.3 of sufficient purity (> 95) such that no further purification is necessary if the deprotection is quantitative. In these cases, the final products will be treated like peptides from solid-phase synthesis: they will be washed with ether, dissolved in water (sometimes with a little cosolvent if necessary to solubilize a hydrophobic sequence), and lyophilized to a powder. In all cases, a small amount of the final lyophilized solid will be characterized by LC-MS with UV detection at 214 nm. Any products < 95% purity as indicated by this method will be purified by semi-preparative HPLC.
The 21 monomers targeted in this specific aim allow for 441 unique oligomer combinations. We will deliver all of these deprotected oligomers in 10-20 mg quantities and greater than 95% purity. We expect to deliver no less than 50 protected oligomers, 10.3, in 10-20 mg quantities and with the same purity criterion. These protected compounds will be selected in part by the availability of the intermediates, though we will perform the coupling reactions at concentrations sufficient to generate all intermediates in amounts sufficient for submission. The protecting groups in 10.3 make these intermediates less exciting for discovery efforts based on our design, and more valuable in reserve for replenishing bioactive oligomers. An analysis of our expectations for this reaction-both the amount of starting materials and reaction yields-is shown in Yields and Delivered Sample Sizes.
Monomer Syntheses and Protection Schemes
The synthesis of 10.1 is shown in Scheme 11. The pyrazines substituted with amino acids, represented by 3.1, will be prepared as shown in our preliminary results (Scheme 3). The acyl imidazole will be opened with a carbobenzyloxy (Cbz) mono-protected diamine, 12.5, that will be synthesized from an amino acid. The product of the ring opening, 11.1, can be deprotected by hydrogenation to give 10.1. These reactions are fast, quantitative, and require only filtration to remove the metal catalyst in order to obtain the desired product in solution. Evaporation and a solvent change will prepare this intermediate for the coupling reactions shown in Scheme 10.
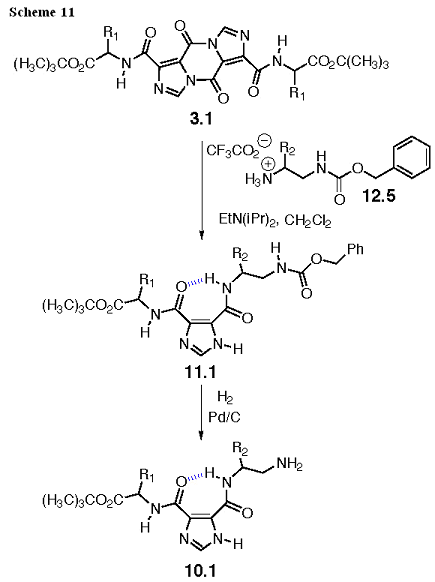
The Cbz protected diamine, 12.5, is prepared from a Boc protected amino acid as shown in Scheme 12. We will use Ala, and Leu amino acids to derive these intermediates, and both have several ester derivatives available commercially. Also, these amino acids do not have side chain functionality to complicate the protecting group strategy or react with the reagents used in the transformation. We will also use a mono-protected ethylenediamine (Gly derivative) commercially available with either Fmoc or Trityl protection, so a protecting group change is all that is needed to obtain this intermediate. The synthesis begins with aminolysis of 12.1 to give carboxamide 12.2, and the amide will be selectively reduced in the presence of the carbamate (urethane) to the amine with boranedimethyl sulfide in tetrahydrofuran (THF) to give 12.3. The amine is protected with the Cbz group to give 12.4, and the Boc group removed with trifluoroacetic acid (TFA) to give 12.5.
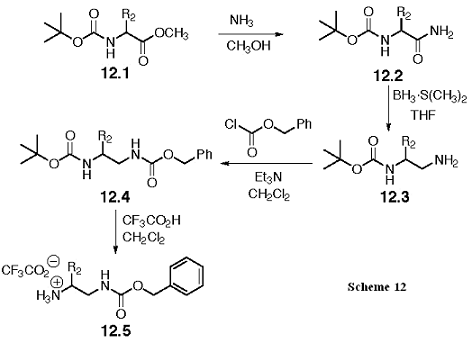
The synthesis of 10.2 is shown in Scheme 13. In contrast with the synthesis to 10.1, the acyl imidazole bond in 3.1, now benzyl ester protected, is opened with a (Boc) mono-protected diamine, 14.5. The product of the ring opening, 13.1, can be deprotected by hydrogenation to give 10.2. As for 10.1, filtration, evaporation, and a solvent change will prepare this intermediate for the coupling reactions in Scheme 10.
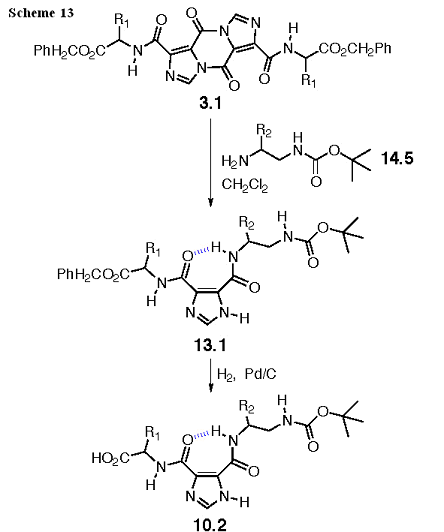
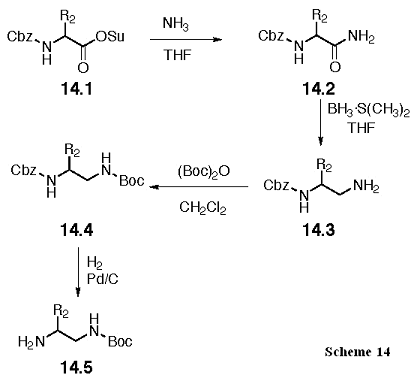
The Boc protected diamine, 14.5, is prepared as shown in Scheme 14. As previously described for 12.5, we will use Ala, and Leu amino acids to derive these intermediates, and the Gly derivative will be made through a protecting group change from a Fmoc analog. Aminolysis will give the carboxamide 14.2, and the amide will be selectively reduced to the amine with borane-dimethyl sulfide in THF to give 14.3. The amine is protected with the Boc group to give 14.4, and the Cbz group removed with catalytic hydrogenation to give 14.5.
Back to Specific Aim 3 Navigation
Back to Design of Library Navigation
Back to the Top
Summary of Library Members
A summary of the targeted library members is given in Table 2 for convenience. The different classes, as well as the compound number, delivered amount, and purity are all given. These are realistic expectations and are detailed in part in Yields and Delivered Sample Sizes.
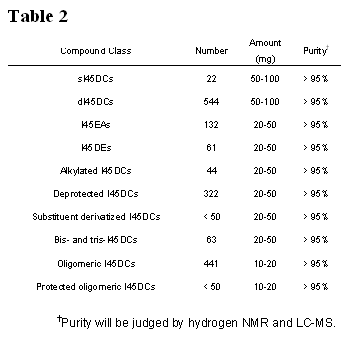
The maximum number of compounds to be delivered over the three years is 1729 compounds. This is a substantial number of compounds, but we have presented the evidence that indicates the varied bioactivity expected throughout the series. Thus, each class has value itself and, in the case of the monomeric I45DA derivatives, is also valuable by comparison with related members. Despite the number of compounds and challenge of their timely production, we have experience with the scaffold and the synthetic transformations are now reliable and uncomplicated. We therefore have full confidence that an organized effort by the team members will succeed in this objective. The acquisition of an automated LC-MS will streamline the characterization of the products.
The synthetic strategy allows for selected intermediates, such as the pyrazines, to be used in the preparation of many compounds (Figure 15). These intermediates are readily prepared on a large scale and require only vacuum filtration to obtain in suitable purity for library synthesis.
A timeline for library development and compound submission is provided below.
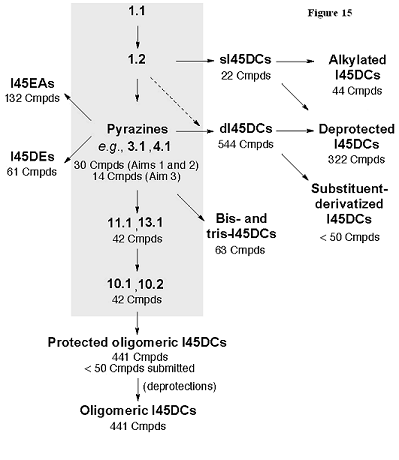
Figure 15. Flowchart showing the synthetic strategy to the different library members. Several common intermediates (highlighted in the gray box) are used and can be prepared in large scale for library synthesis.
Back to the Top
Yields and Delivered Sample Sizes
The RFA cites a minimum of 10 mg to be delivered for each library member, representing 20 µmol for a 500 g/mol I45DC (an average-to-high MW for the monomeric I45DCs). Nonetheless, we have targeted greater sample sizes, as it is expected that these libraries will be most valuable if they can be tested in a vast array of bioassays. It is also easy to provide more of each compound when requested, but our concern is that if a given sample would be consumed before its bioactivity is discovered, the request to replenish the stock may not be made. In addition, some compounds simply help complete the SAR for a structural class, and therefore even inactive or weakly active members should be available in large amounds for testing in a variety of bioassays.
The I45DA starting material is commercially available at $80.20 for 25 grams (Aldrich Chemical Co.). There is also an Organic Syntheses preparation from inexpensive and readily available tartartic acids. Thus, generating the pyrazine intermediates for our libraries will not be limited by access to the starting material. The monomeric I45DCs, I45EAs, and I45DEs are generally made in three synthetic steps. The exceptions are the sI45DCs made in two steps and the 36 unique dI45DCs made in four steps. A simple calculation diagramed in Figure 16 supports our premise that the targeted amounts and expectations are quite realistic. The goal for dI45DCs is to submit 100 mg per sample. In Figure 16, the product 3.2 is considered to be a 400 g/mol library member with an amino acid ester substituent and primary amine. Thus, 100 mg represents 0.25 mmol. The calculation allows for a poor yield (50%) obtained in every step along the reaction path (this is a yield occasionally obtained in one step, but
not in every step to the examples we made to date). Working backwards, we would then need to start with 2.0 mmol of I45DA for each such library member. Using this as the estimate for all of the 1175 monomeric compounds indicates that we would need a maximum of 361 g of the I45DA starting material. No problems are expected in obtaining this commercially, as my group has likely experimented with a similar amount in recent years. The reaction of 1.1 to give pyrazine 1.2 has been run on a 15 gram scale and requires only vacuum filtration in order to collect the product at the end of the reaction. It is expected that increasing this reaction to at least 25 grams is possible without complications. We do not expect to go to even greater scales, as the intermediate is best prepared immediately before its use due to its reactivity.
Back to the Top
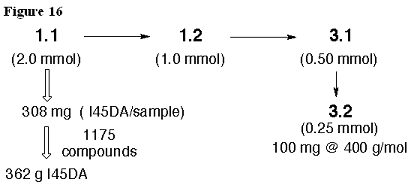
Figure 16. Analysis of sample sizes in monomeric I45DA derivatives. This example seeks 100 mg of a 400 g/mol library member and allows for a poor yield (50%) in the steps leading to this compound.
A similar analysis is shown in Figure 17 for an oligomeric I45DC. The scale of subsequent reactions will vary depending upon the yields we obtain in early library synthesis, yet these calculations assist us in selecting proper reaction scales for a given library.
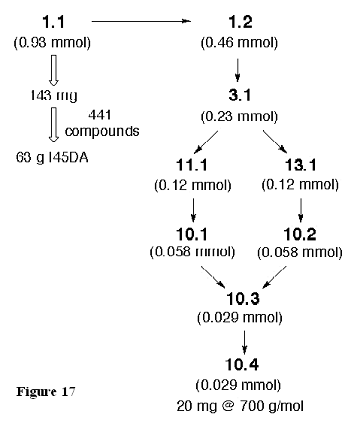
Figure 17. Analysis of sample sizes in oligomeric I45DA derivatives. This example seeks 20 mg of a 700 g/mol library member and assumes a poor yield (50%) in the steps leading to the penultimate intermediate 10.3.
This linear sequence totals six chemical steps from 1.1. However, this synthetic pathway does not hinder the production of large numbers of compounds because the libraries are synthesized late in the sequence, and late intermediates can be prepared on a large scale. For example, pyrazines 3.1 have been prepared by multigram scale reactions and can be crystallized before their use in the production of the combinatorial libraries. The smaller libraries of 11.1 and 13.1 can be synthesized and purified in parallel (21 compounds each). Likewise, deprotection of the Cbz group in 11.1 and benzyl ester of 13.1 can be done in parallel, and we expect only filtration will be required to yield the libraries of intermediates 10.1 and 10.2, respectively. This is a reasonable number of syntheses to generate the intermediates for the 441-member library resulting from the combination of these products. The amide bond coupling reactions and extractions are performed with an automated liquid handling system. The resulting products, 10.3, are expected to be of high purity from the extraction alone, although they can be further purified as necessary by solid-phase extraction on silica gel in parallel to give the products in 95% purity or greater. The final deprotection is expected to yield products with equivalent purity after lyophilization. Thus, only in the last two steps of this sequence are we handling larger numbers of compounds, and even then the procedures for purification and submission are straightforward.
Second Generation Libraries
The roughly 1700 compounds in this proposal are but a fraction of the possible structures of new I45DA derived compounds, and each class can be greatly expanded. The chemistry to make these analogs for the purpose of generating detailed structure-activity relationships is no more difficult than those proposed herein. In addition, the starting materials to these compounds are available commercially and in diverse structures themselves (amines, alcohols, anilines, heterocyclic amines, and amino acid esters). Thus, biologically active members of the primary libraries can be readily expanded by analog synthesis and bioassay, thereby speeding up discovery and understanding of biological processes manipulated by the I45DA derivatives. In some cases, subtle modification of a bioactive parent can generate functionality that is used in the generation of secondary libraries. To illustrate this is part of our second specific aim.
Back to the Top
Sample Handling During Synthesis
Shown in the preliminary results is a model library (Oligomer Library Synthesis and Purification: A Model Scale-Up) prepared in a 10-day period by the PI. We will optimize our methods for the efficient preparation of the compounds in this proposal. An Argonaut Quest 210 Synthesizer is equipped for solid phase or solution phase organic synthesis. The Tecan Genesis RSP 100 Combinatorial Synthesizer is equipped for organic synthesis and has four robotic probes with interchangeable septa piercing co-axial (for inert gases) needles or disposable tips or both. Both tip types are conducting and therefore have the capability to do on deck extractions of aqueous/organic phases. The synthesizer is equipped with several different rack systems. These racks hold a variety of reaction vessels, including microtiter plates, test tubes and scintillation vials (each type with open and septa sealed configurations).
The system is equipped with on deck and off deck reaction processing for heating, cooling and agitation. John DiCesare is an experienced user of these two
instruments and will oversee the production of the libraries with this equipment. Reactions with the most chemically sensitive pyrazines such as 1.2 will be done on an individual basis under an inert gas. In addition, the aniline-substituted pyrazines, represented by 4.1, are often insoluble in our reaction solvents, only dissolving as the reaction proceeds to give soluble product. We can weigh out and distribute these pyrazines to individual reaction vials if necessary, but we will also explore how much variation there is in weight by measuring a volume of an evenly divided powder (possibly with a diluent). The pyrazine or the amine can be used in excess and the silica gel chromatography used to purify the product will still yield clean material for submission.
Reactions will generally be done in glass vials and magnetically stirred or gently shaken depending upon the sample size. The reactions of pyrazines with hindered or less reactive amines or amino acids take 12-18 hours at room temperature. We expect to use these conditions for the libraries. We will change conditions as deemed necessary to optimize the transformation. We will have to heat the reactions that open the acyl imidazole bond with alcohols (reflux in THF). Aliquots of selected reactions will be analyzed by LC-MS with detection at 214 nm in order to determine if the reactions are complete prior to purification. We expect this information will be a valuable addition to our final database, and in solving problems in the unlikely event that purification does not yield all of the desired products or if select products are obtained in unexpectedly low yield.
We will run library syntheses in groups of 20-100 members, making our decision based on the best combinations for the given class. The intention is to always maintain a very manageable number of reactions in progress, such that a flow is established through the different stages of preparation: namely, intermediate pyrazine synthesis, library reaction synthesis, solid-phase extraction on silica gel, sample evaporations, characterization of the product, and preparation for delivery to the NIH repository. Establishing a "schedule" will allow for efficient production of the libraries. A bar code printer and reader will be used to keep track of library members through the various steps of the above cycle, and to cross-reference analytical data with both archived and submitted samples. Labels for vials will be generated in advance of the experiments. Library members will be added to a Microsoft Excel file with a code signifying the class of structure and its member number.
This organization process will also help the group see the entire project with clarity, so that tasks toward the objective can be met in a timely fashion.
Back to the Top
Solid-Phase Extraction
We have used selective solubilization of impurities, crystallization of the desired product, and solid-phase extraction on silica gel to purify I45DA derivatives in our preliminary results (Baures, 2002; Wiznycia, 2002; Wiznycia, 2004)*. The optimal and fastest method is solid-phase extraction on silica gel, and this will be the primary purification method for all of the compounds in specific aims 1, 2, and 3. We will purchase solid-phase extraction columns of various sizes in order to facilitate the rapid purification of compounds.
The most prevalent impurities in the monomeric I45DA libraries result from incomplete reactions or from competition by hydrolysis, leading to products such as imidazole-4-carboxamide-5-carboxylic acid, or the like. These byproducts are significantly more polar than our desired I45DA derivatives. The silica gel columns are executed as a filtration, with little worry for separating closely eluting compounds. Vacuum manifolds are available and will help in the rapid collection of the product.
In the model library, the columns were first wet with ethyl acetate. Gentle vacuum was drawn to both further pack the column and draw through some of the solvent. A 1:1 mixture of ethyl acetate/hexane was then drawn through before the samples were loaded. The eluant polarity was increased as fractions were collected in test tubes. In our experience, the compounds elute either late in the ethyl acetate fractions or early in the ethyl acetate/methanol fractions. This is also consistent with the solid-phase extraction conditions used for the antiprolerative I45DCs (Perchellet, 2005)*, although the separation of fractions was not required for these compounds. We expect this method will work for the libraries herein, and this will be the starting point for our purifications.
Evaporation of the eluted samples can be readily done under a stream of N2 gas or with the speed-vac concentrator systems (two of each available at TU). The later are equipped with several rotor types, including those to hold scintillation vials, test tubes, and microtiter plates. Each system has an independent vacuum pump and cold trap.
Back to Purification and Characterization Navigation
Back to the Top
Extractions
The protected oligomers, 10.3, in specific aim 3 will be purified first by extraction. The earlier intermediates, 11.1 and 13.1, will be purified by solid-phase extraction to analytical purity as determined 1H NMR spectroscopy and LC-MS with UV detection at 214 nm. Hydrogenation is generally a clean reaction and requires only filtration, though a 1H NMR spectrum will be collected on 10.1 and 10.2 to ensure this is the case.
The coupling reaction with the water-soluble carbodiimide is expected to go in high-yield, as the carboxylic acid and amine are both unhindered and freely reactive (a main exception to high yields in this reaction). These coupling reactions will be done in ethyl acetate and, at the end of the reaction, we will wash the product in the reaction vial itself. The ethyl acetate will remain the top layer and the aqueous washes can be removed by using the robotic liquid handling equipment for the extractions.
We will wash with 10% citric acid, water, 1 M NaHCO3, water, and brine. The remaining organic layer can be dried with anhydrous MgSO4 and filtered. The products, 10.3, will be flashed through a pad of silica gel to remove unreacted imidazole starting materials (10.1 and 10.2) as well as possible inorganics. They will be analyzed by LC-MS with detection at 214 nm.
Back to Purification and Characterization Navigation
Back to the Top
Product Characterization
The products from all specific aims will be characterized by 1H NMR spectroscopy and LC-MS with detection at 214 nm. The analytical data will accompany each deposited compound and be made readily available to other researchers using these compounds.
Back to Purification and Characterization Navigation
Back to the Top
Efficient Sample Handling
We will prepare for the characterization process by separating the product into two weighed and labeled vials. A majority of the material will be transferred to one vial, with the other vial containing a smaller fraction of the total. Both of the vials will be dried to a constant weight before determining the total amount of product. The major amount of each product will be submitted to the small molecule repository, while the smaller sample will be used for characterization and as a reserve sample.
Back to Purification and Characterization Navigation
Back to the Top
1H-NMR Spectroscopy
The smaller sample will be dissolved in CDCl3 in order to collect the 1H NMR spectrum. The weight of the vial will allow us to determine the concentration of the sample. This is important since we know that the amide and imidazole NHs in the NMR spectrum of these products is dependent upon concentration. Thus, we will be able to report the exact concentration in the documentation provided with the library. The spectra will be examined for its consistency with the expected structure. We will provide a copy of each spectrum with the submitted samples, and also make the spectrum available to others in the supporting information during publication.
Back to Purification and Characterization Navigation
Back to the Top
LC-MS Analysis
We have previously used HPLC to determine homogeneity of products (Wiznycia, 2004; Perchellet, 2005)*. Following these examples, the samples will be injected on an analytical C18 column and then eluted with a gradient of CH3CN/H2O.
The gradient begins with a 1:1 concentration of CH3CN/H2O and steps to 95:5 CH3CN/H2O over 14 min., followed by pure CH3CN at 15 min. The run ends at 20 min. and returned to the 1:1 ratio. The utility of this method was the baseline separation of two homologs (one carbon difference in a bis-I45DC weighing approximately 700 g/mol) that we reported recently (Wiznycia, 2004)*. The samples will be detected at 214 nm. The oligomers in specific aim 3 should elute satisfactorily with this method, though we can certainly make modifications if necessary.
We will again use the baseline separation of similar compounds as a test of the suitability of the method. These conditions will be used as a starting point for setting up the LC-MS instrument.
The LC-MS will also determine the mass spectrum of the compound in order to provide the parent ion in support of the molecular structure. In most cases the parent ion is expected to be the dominant peak and no analysis of the secondary ions will be done. We expect to purchase an LC-MS utilizing one of the softer ionization methods. We will select between these ionization choices by comparing the results obtained with commercial instruments for representative compounds in our current library. Nonetheless, it is possible that certain derivatives in one or more library will fragment too rapidly and lead to weak parent ions.
A collaboration with Elizabeth Stemmler at Bowdoin College is ongoing in order to determine the mechanisms of decomposition in the mass spectrometer for compounds represented by 4.2. The compounds analyzed to date have not been difficult to identify (the goal in this collaboration is to study the influence of the imidazole on the ionization mechanism, and we have noted
varying ratios of a-, b-, and y-type ions in comparison with the parent peptides). This work has also shown us that the sodium salts of the I45DCs give strong and stable parent ions in the mass spectrometer.
On the other hand, the larger oligomers from specific aim 3 may require MALDI, FAB, or ESI to satisfactorily characterize the parent ions. These techniques are available at nearby campuses in Oklahoma (OSU and OU) and Kansas (KU). I have previously used the KU MS facility for service with the bis-I45DCs, so this will again be used if necessary. This service can run as much as $30/sample, but it is not anticipated that all of the compounds in specific aim 3 will need to be analyzed by this method.
Back to Purification and Characterization Navigation
Back to the Top
Sample Storage and Archival Of Analytical Data
The samples of all I45DA derivatives to date are stable materials at room temperature. Most are solids, although a few amino acid derivatives have formed thick oils or glasses. We expect that these compounds could be lyophilized to produce powders for ease of handling, storage, and for submission to the NIH MLSMR.
Samples of each final compound in this proposal will be saved for potential future use, either for use in further confirming structure or biological activity or both. These samples will be stored in vials under an inert gas at room temperature in the dark. The vials will be organized in cardboard freezer boxes by both library type and substituents of the members. We will code the vials by the library and compound, recording this in the Excel file used for tracking the entire library. All analytical data will be stored in acid-free plastic sleeves in three-ring binders.
The binders will be organized to coincide with the boxes, such that compound and data retrieval is straightforward. This analytical data will also accompany the compounds submitted to the repository.
In addition to the final compounds, an analytical sample of every intermediate beyond 1.2 will be saved. In some cases, we will reserve quantities of intermediates produced in the project and expected to be of value in the production of secondary libraries.
The long-term molecular stability is not a concern with these compounds. All samples prepared to date have been stored as solids at room temperature with no discoloration or decomposition. The functional groups in these compounds and in the proposed libraries are all strong and inherently quite stable.
Moreover, the intramolecular hydrogen bond makes the amides of the I45DCs even less reactive than amides in general. For example, we determined the pKa of model compounds by following the C2-H with 1H NMR spectroscopy as a function of pH (Rush, 2005)*. In this work we observed neither changes in chemical shifts nor additional signals that might suggest partial amide bond hydrolysis-even in our aqueous solutions at pH ≈ 1.
To facilitate the use of the libraries by other researchers, we will create and maintain a web page where access to the library information can be obtained. This is described in greater detail below.
Back to the Top
Public Dissemination
We have reported some of the synthetic methods needed to produce the I45DA based compounds in the libraries in this proposal (Baures, 2002; Wiznycia, 2002; Wiznycia, 2004)*. New methods or substantial improvements to the synthetic procedures will be reported through one of the organic chemistry journals. The graduate students will develop the methods for the oligomer synthesis as his or her thesis projects, and this will also be reported in one of the organic chemistry journals.
We will report the preparation, purification, and characterization of the libraries. This will be sent to The Journal of Combinatorial Chemistry, although other chemistry journals could also be appropriate choices. Here we will separate the libraries much as the proposal has separated them. Thus, the libraries in specific aims 1 and 2 that are monomeric in I45DA will be described separately from the libraries generated under specific aim 3 that are oligomeric in I45DA. In addition to differences in the number of I45DAs above, these libraries are also designed with different hypotheses and expected targets in mind. These reports will provide details pertinent to the production of these specific libraries, as well as for the generation of secondary libraries. For instance, we will describe sample handling, reaction times and conditions, qualitative analysis results of the reactions (TLC or LC-MS or both), and how each sample was purified. The analytical data will also be included as supplementary
information.
A web page will be created and maintained in order to give quick access to general and specific chemical structures in the library, analytical data, and procedures for the preparation of a compound in the specific class. We will also link to references (abstracts or papers depending upon the work) of the chemistry, conformational analysis, and biological activity of I45DA derivatives. This will be a quick source of information for the researchers working with biologically active I45DA derivatives identified in this proposal. We think this method of sharing our data is the most likely to provide widespread access to the information for researchers. Moreover, the nested web pages and data can be made available on CDs for researchers either lacking or with poor internet connectivity.
Back to the Top
Timeline of Proposed Work
The synthesis, purification, characterization, and delivery of the roughly 1700 compounds for the NIH repository will take the entire three years of the grant. A timeline for the preparation of the libraries over the three years is shown in Table 3. This timeline estimates the quarters spent in the synthesis and purification and characterization of the libraries for each of the three specific aims. In addition, we also estimate the quarters during which delivery of libraries can be made.

The library deliveries are rather evenly split out over the three years, with 451 compounds delivered in year 1 and 669 compounds in year 2 (with 322 of these being simple deprotections of earlier compounds). The remaining 500 or so compounds will be delivered in year 3.
The workload is considered balanced on the basis of the expected efforts required for the delivery of these libraries. It was our design to target the easier monomeric I45DA based compounds first, as this will help meet our early goals and develop experience with these derivatives for the new group members. The more difficult monomeric derivatives, like the dI45DCs with primary alkanamines, are submitted in a later batch. In addition, the amino acid ester derivatives are saved for the last batch of monomeric derivatives, as work on the oligomeric I45DCs will already have generated some pyrazines with amino acid esters. A few of these intermediates (3.1) are in common between these libraries in the different specific aims.
Back to the Top
Project Management
The PI has experience in combinatorial chemistry from time spent at Signature BioScience (Signature BioScience, Inc., 2003)*. This company maintained a large library of chemical compounds for testing in their proprietary screens (Cytection, 2001)*. The structures of the compounds, their bioassay data against multiple targets, and some of their physical property data were kept in a company database. The PI was selected as a project manager while there and led in the design of libraries specific to his project, as well as in the selection of compounds for informer libraries to be used in future drug discovery efforts by the company.
A brief synopsis of the history of Signature BioScience is warranted in order to place the PI's experience in context. Signature BioScience started as an instrument company, but morphed into an anticancer drug discovery startup around 2001 under the direction of Mark McDade (in 2006 CEO of Protein Design Labs). This transformation was facilitated by the purchase of PrimeCyte, Inc. (PrimeCyte, Inc., 2002)*, a bioassay platform out of Seattle, and two groups of chemists in the San Francisco Bay area. The chemists were a combination of the small molecule group from Protein Design Labs (Protein Design Labs, 2001)* and Cambridge Discovery Chemistry, Inc., (Cambridge Discovery Chemical, 2001)* who were owned at the time by Millenium Pharmaceuticals, Inc. The chemists inherited a major agrochemical research facility. Located on-site were over forty thousand fine chemicals, intermediates, and products from years of agrochemical research. A large service analytical lab with two LC-MS systems for walk-up use and a 400 MHz NMR spectrometer was also available. In addition, the company had its own robotic lab for the preparation of combinatorial libraries, robotic liquid handling equipment for high-throughput purification of the compounds, and preparative HPLC systems. The company employed over 100 people at its height (approximately 30 chemists) and had a verbal promise of $40 million in venture startup for advancing the objectives of anticancer drug discovery at the time of the PI's hire. When this commitment was withdrawn, a bridging loan of $10 million was promised to continue drug discovery at a smaller scale, but this too was unexpectedly voided.
The PI's time spent at Signature BioScience, albeit brief, provided significant real-world experience in the use of combinatorial chemistry for discovery purposes in biological systems. In addition, the use of the company databases and archival system illustrated to the PI the manner in which large volumes of information must be organized.
John DiCesare has combinatorial chemistry experience from time spent at Research Triangle Institute. He has most recently used these techniques and the equipment available at The University of Tulsa toward the discovery of nerve agent sensors. His experience will be valuable in meeting the objectives of this grant by directing the use of the automated synthesizers and liquid handling equipment.
Back to the Top